Ottobre è il mese di sensibilizzazione sulla sindrome di Joubert e l’occasione per raccontare un progetto di ricerca di Fondazione Telethon su questa malattia, i cui risultati potrebbero rivelarsi utili anche per altre malattie genetiche rare.
«La prima cosa che ci chiedono i genitori quando il loro bambino o la loro bambina riceve la diagnosi di sindrome di Joubert è come andranno le cose» racconta Enza Maria Valente, professoressa di genetica medica all’Università di Pavia e responsabile del laboratorio di diagnostica genetica e del Centro di ricerca di neurogenetica dell’IRCCS Fondazione Mondino, sempre a Pavia.
La sindrome, caratterizzata dalla malformazione congenita di alcune strutture poste alla base del cervello, è infatti molto eterogenea, con alcuni pazienti che riescono a condurre una vita quasi normale e altri che vanno incontro a disabilità intellettiva e disturbi anche molto gravi di vari organi. «I genitori vorrebbero sapere se il figlio avrà problemi alla vista, se e quando svilupperà insufficienza renale, se avrà una disabilità intellettiva e quanto sarà grave. Non sempre però riusciamo a rispondere, perché la sindrome di Joubert è molto complessa non solo nelle manifestazioni ma anche negli aspetti biologici». Serve ancora tanta ricerca per conoscerla meglio.
Una malattia delle ciglia (cellulari)
«Nel 2003 ero appena tornata da Londra, dove avevo finito il dottorato di ricerca in neurogenetica, e lavoravo all’Istituto CSS-Mendel di Roma. Un giorno arrivò dalla Sicilia una famiglia con un bambino con questa malattia praticamente sconosciuta e il professor Bruno Dallapiccola, allora direttore scientifico dell’Istituto, mi chiese se mi interessava studiarla. Da allora non ho mai smesso di fare ricerca sulla sindrome di Joubert e non è mai venuto meno l’entusiasmo di farlo» ricorda Valente, che su questa malattia ha lavorato a lungo - e continua a farlo - con progetti di ricerca finanziati da Fondazione Telethon, diventandone un riferimento internazionale. «Quando ho cominciato a occuparmene non se ne sapeva quasi niente, neppure quali fossero i geni coinvolti» dice.
Oggi certamente ne sappiamo di più. «Per esempio, che c’è almeno una quarantina di geni che, quando mutati, possono dare origine alla malattia. E che la categoria generale alla quale appartiene è quella delle ciliopatie». Sono malattie causate da alterazioni delle cilia primarie, strutture che appunto hanno la forma di un ciglio che si protende dalla superficie di moltissime cellule. Negli anni abbiamo scoperto che le cilia svolgono funzioni essenziali per lo sviluppo del sistema nervoso e di altre strutture nella vita embrionale, ma anche per la normale fisiologia dell’organismo, ad esempio per il corretto funzionamento di cellule della retina, dei reni e del fegato.
Il cuore sempre oltre l’ostacolo
«Le questione aperte sulla sindrome di Joubert, però, sono ancora tantissime» sottolinea Valente con il sorriso aperto che la contraddistingue. «La ricerca - commenta - è una grande sfida, ma è anche fonte di grandissime soddisfazioni. Ogni esperimento “venuto bene”, ogni nuova idea da mettere in pratica, ogni scoperta che facciamo, grande o piccola che sia, rinnova ogni giorno l’entusiasmo che ci anima come ricercatori e che ci spinge ad andare avanti e ad affrontare anche i momenti difficili ed i fallimenti (che non mancano) con un’attitudine positiva. Fare ricerca è uno dei lavori più belli del mondo, perché è sempre nuovo ogni giorno, c’è sempre un nuovo obiettivo da raggiungere, una nuova sfida in cui lanciare “il cuore oltre l’ostacolo” e nuovi giovani a cui trasmettere questa passione. Per non parlare del fatto che riuscire a dare un aiuto concreto ai piccoli pazienti e alle loro famiglie, fornire risposte che prima non c’erano, cambiare in qualche modo le loro vite, è davvero impagabile».
Casi senza diagnosi
Una delle grandi questioni aperte è il fatto che nel 30-40% dei casi non si riesce ad arrivare a una diagnosi genetica certa. «Negli ultimi anni sono stati identificati molti nuovi geni responsabili della sindrome di Joubert grazie all’avanzamento delle tecnologie di sequenziamento del Dna, con la possibilità di sequenziare in tempi brevi e a costi contenuti l’insieme delle porzioni dei nostri geni che codificano per le proteine (in gergo tecnico, l’esoma). Per molti pazienti, però, il sequenziamento del “solo” esoma non offre risposte». Ed è proprio questo uno dei punti sui quali intende intervenire il nuovo progetto di ricerca di Valente. «L’ipotesi è che siano coinvolte mutazioni che non possiamo vedere con le tecniche convenzionali, per quanto avanzate, per esempio perché riguardano regioni del nostro dna che, pur essendo “non codificanti”, possono avere importantissime funzioni regolatorie ancora largamente sconosciute. Per cercarle dobbiamo guardare con altre tecniche “oltre la genetica convenzionale” e vedere se in questo nuovo territorio si trova quanto manca».
Modelli cellulari super personalizzati
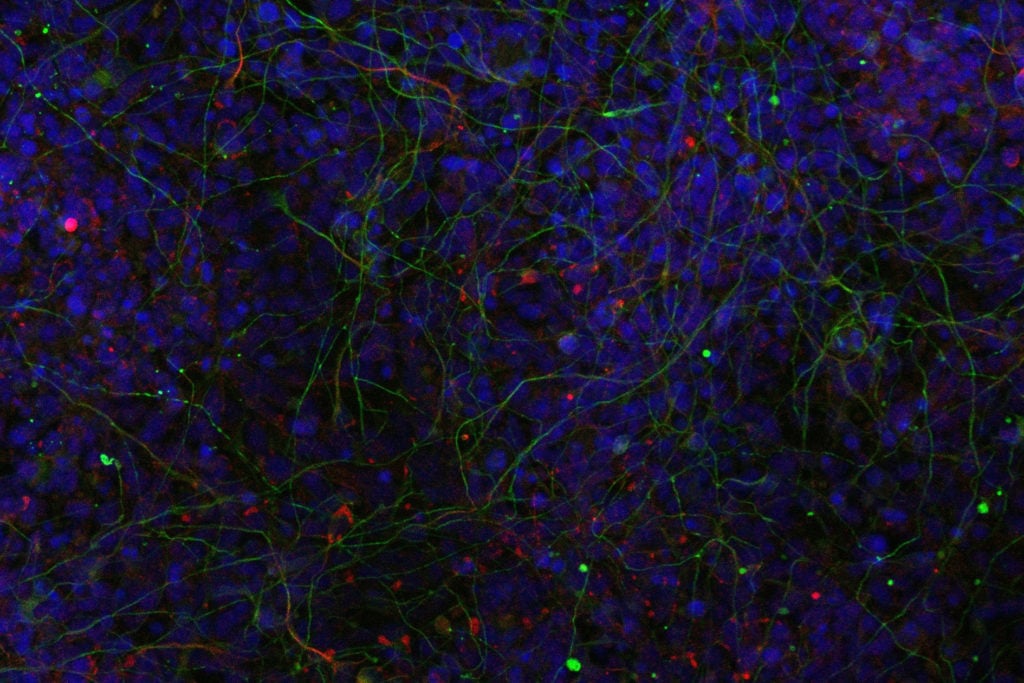
Un altro quesito importantissimo e ancora senza risposta riguarda il fatto che mutazioni nello stesso gene possono provocare manifestazioni cliniche molto differenti, con il variabile coinvolgimento di organi diversi. «Vogliamo capire meglio come funzionano le diverse varianti di uno stesso gene, perché le mutazioni non sono tutte uguali. Per farlo, utilizzeremo un modello cellulare molto promettente, le cellule staminali pluripotenti indotte ottenute da pazienti portatori di specifiche mutazioni. Abbiamo in programma di differenziare queste cellule nei tessuti specifici interessati dalla malattia (ad esempio in cellule neuronali o renali o precursori dello scheletro), per scoprire se la stessa mutazione può avere un impatto differente sulle funzioni cellulari a seconda del tipo di tessuto».
Secondo Valente, questo tipo di modelli cellulari aiuterà a capire meglio gli effetti delle mutazioni coinvolte nella sindrome di Joubert, permettendo in futuro (si spera) di dare prospettive più realistiche a quei genitori che cercano di immaginare il futuro dei loro bambini. Inoltre, questo stesso approccio potrebbe rivelarsi utilissimo anche per studiare altre malattie genetiche rare geneticamente complesse, come malattie del neurosviluppo, epilessie o disabilità intellettive. E strada facendo, impareremo sempre più dettagli su come funziona il nostro organismo o, in altre parole, sui meccanismi che ci rendono davvero quello che siamo: umani.