Angela Gritti dell’Istituto San Raffaele-Telethon di Milano racconta come si sta cercando di applicare la terapia genica per uno degli organi più difficilmente accessibili del nostro organismo.
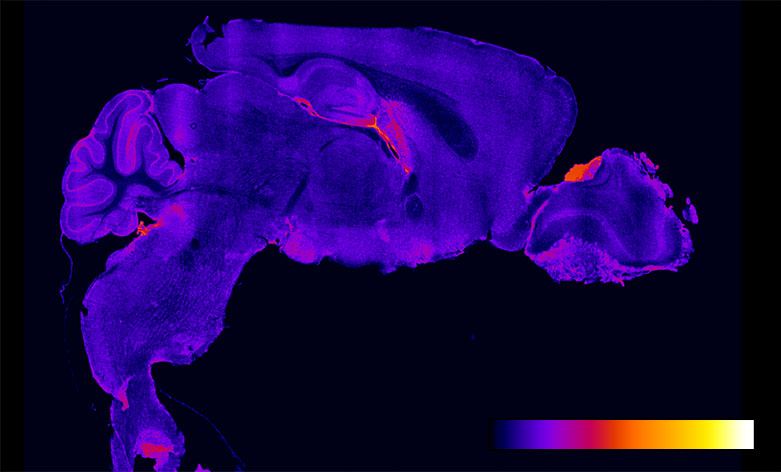
Distribuzione degli accumuli del ganglioside GM2 nel cervello di un topo adulto normale. Scala colore: Blu: no accumulo; rosso/arancione/giallo: accumulo crescente 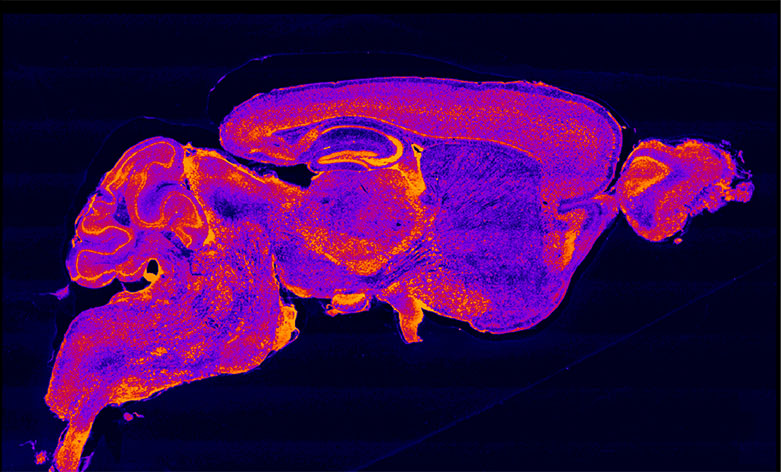
Distribuzione degli accumuli del ganglioside GM2 nel cervello di un topo Sandhoff non trattato negli ultimi stadi della malattia. Scala colore: Blu: no accumulo; rosso/arancione/giallo: accumulo crescente 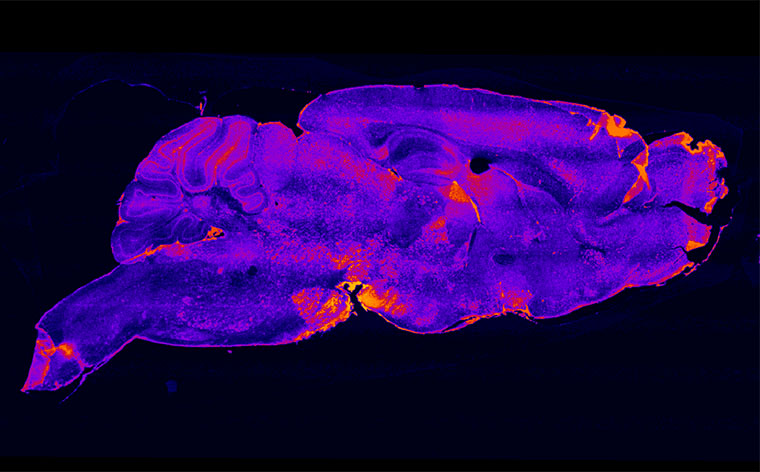
Distribuzione degli accumuli del ganglioside GM2 nel cervello di un topo Sandhoff due mesi dopo il trattamento combinato con iniezione di vettore intracerebrale e trapianto di midollo. Scala colore: Blu: no accumulo; rosso/arancione/giallo: accumulo crescente
1 / 3
La terapia genica è ormai una realtà terapeutica per diverse malattie genetiche, anche grazie all’importante contributo della Fondazione Telethon. Grazie ai progressi nell’ingegneria genetica e nella conoscenza dei meccanismi di base oggi siamo in grado di trasformare alcuni virus in trasportatori di Dna terapeutico e fornire così una versione funzionante di geni altrimenti difettosi e responsabili di specifiche malattie.
Tuttavia non tutti i tessuti – e quindi non tutte le malattie – sono ugualmente affrontabili con la terapia genica: tra le sfide più grandi c’è indubbiamente quella di raggiungere il cervello, organo dotato di una barriera naturale e quindi difficilmente accessibile.
«Trovare strategie in grado di raggiungere il cervello può fare da apripista anche per altre malattie: per questo è importante continuare a sostenere la ricerca, come fa Fondazione Telethon»
Angela Gritti, ricercatrice SR-Tiget
All’Istituto San Raffaele-Telethon per la terapia genica (SR-Tiget) di Milano, Angela Gritti studia da molti anni come applicare la terapia genica a malattie genetiche del sistema nervoso che si manifestano già nei primi anni di vita e per cui ad oggi, nonostante gli sforzi, non esistono cure. «Si tratta tipicamente di malattie dovute all’accumulo di prodotti del metabolismo, che non vengono smaltiti e diventano tossici per le cellule nervose. Alla base c’è spesso un difetto enzimatico: a seconda della proteina carente si accumula una specifica sostanza che danneggia il cervello. È il caso delle leucodistrofie e delle gangliosidosi: ogni malattia ha la sua peculiarità, ma le caratteristiche comuni sono drammatiche. Dopo un esordio che può essere anche nei primi anni di vita, si assiste alla perdita progressiva delle capacità motorie e cognitive, fino purtroppo alla morte precoce».
Un caso di successo
Un caso brillante di successo della terapia genica per una malattia genetica di questo tipo è frutto proprio della ricerca dell’SR-Tiget: si tratta di Libmeldy, per il trattamento della leucodistrofia metacromatica, che alla fine del 2020 è stata approvata come farmaco nell’Unione europea. È una terapia genica ex-vivo, in cui le cellule del paziente vengono ingegnerizzate al di fuori dell’organismo e reinfuse dopo la correzione con il vettore virale. Il vettore è di tipo lentivirale, deriva cioè dal virus Hiv ed è in grado di integrarsi nel Dna della cellula ospite, permettendone quindi una correzione stabile e duratura. Un aspetto poco intuitivo ma cruciale è che a essere corrette con la terapia non sono le cellule direttamente colpite dalla malattia, i neuroni, ma le staminali ematopoietiche, che si trovano nel midollo osseo e danno origine a tutti gli elementi del sangue. Alcune delle “figlie” di queste cellule, infatti, vengono richiamate nel cervello proprio a causa del processo infiammatorio provocato dall’accumulo di sostanze tossiche: grazie alla capacità di produrre quantità di enzima anche superiori ai livelli normali e di secernerlo sono in grado di detossificare il cervello e preservarlo dalla degenerazione, purché l’intervento sia effettuato nella fase pre-sintomatica, quando il danno non è ancora irreversibile.
Questo approccio, che sta dando risultati molto promettenti anche per un’altra malattia genetica da accumulo (la sindrome di Hurler, o mucopolisaccaridosi di tipo 1H), non funziona però per tutte le malattie di questo tipo.
Come spiega Angela Gritti «molto dipende da struttura, dimensioni e modalità di funzionamento del gene-malattia e della proteina che codifica. Per esempio nel caso delle gangliosidosi, caratterizzate dall’accumulo nel cervello di particolari grassi chiamati sfingolipidi, questo metodo non funziona, né finora si è trovato un modo efficiente e sicuro per fornire al cervello l’enzima mancante in quantità sufficiente e nei tempi giusti. Le forme più gravi di queste malattie hanno infatti un decorso molto rapido e lo stesso trapianto di cellule staminali non riesce a ripristinare la produzione della proteina in tempo. Da anni stiamo quindi studiando una strategia combinata, che al trapianto affianchi una somministrazione di enzima direttamente nel cervello, tramite una terapia genica in vivo questa volta».
I risultati della strategia combinata
In un articolo appena pubblicato su Molecular Therapy - Methods & Clinical Development*, Gritti e il suo gruppo hanno dimostrato nel modello animale della malattia di Sandhoff, un tipo di gangliosidosi, che la strategia combinata offre un netto vantaggio rispetto ai singoli trattamenti nel fornire rapidamente e stabilmente l’enzima mancante al sistema nervoso sia centrale che periferico. «I topi così trattati, che riproducono i sintomi della malattia umana, vivono di più e meglio e presentano sia un aumento dell’attività enzimatica sia una riduzione dei metaboliti tossici nel cervello – commenta Gritti. La terapia genica in vivo garantisce una disponibilità di enzima quasi immediata e dà il tempo alle cellule staminali ematopoietiche trapiantate di arrivare al cervello, attecchire, e iniziare a funzionare, esercitando anche la loro azione antinfiammatoria: per una malattia dal decorso rapido come questa si tratta di un aspetto cruciale. Inoltre, abbiamo individuato la soglia di attività enzimatica (misurata nel liquido cerebro-spinale) necessaria per avere un impatto significativo sulla patologia, che deve essere almeno il 20-25 per cento di quella fisiologica».
Questo studio offre quindi un contributo importante allo sviluppo futuro di terapie combinate per il trattamento delle gangliosidosi, nonché per interpretare i risultati delle terapie sperimentali in corso. Da poco sono stati infatti pubblicati su Nature Medicine i risultati preliminari di uno studio clinico di terapia genica in vivo per la malattia di Tay-Sachs, un’altra forma di gangliosidosi dal decorso analogo a quella di Sandhoff e anch’essa dovuta deficit dell’enzima esosaminidasi (a differenziare le malattie è la porzione dell’enzima coinvolta, rispettivamente la subunità alfa e la beta). L’approccio utilizzato è una terapia genica in vivo basata su vettori diversi di tipo AAV, che a differenza di quelli lentivirali non si integrano nel Dna, iniettati in una particolare area del cervello e nel liquido cerebro-spinale.
«I risultati sono ancora molto preliminari – commenta Angela Gritti. Da quanto riportano gli autori la terapia per ora è stata ben tollerata, ma serve più tempo per capire se è realmente in grado di cambiare la storia naturale di questa grave malattia. Un aspetto cruciale, che con il nostro approccio speriamo di superare, è di aumentare la quantità di vettore che raggiunge realmente le cellule del cervello, e quindi di enzima funzionante che si riesce a produrre. A complicare le cose c’è anche il fatto che l’enzima è costituito da due subunità, codificate da due geni diversi: dobbiamo quindi fornire non uno, bensì due geni, e nelle proporzioni corrette. Questo si può fare somministrando due vettori diversi, oppure con un unico vettore contenente i geni per entrambe le porzioni dell’enzima: i prossimi esperimenti ci diranno quale possa essere la strategia migliore. Di fronte a malattie così gravi e complesse è un buon segno il fatto che ci siano più approcci terapeutici in fase di studio. Inoltre, trovare strategie in grado di raggiungere efficacemente il cervello può fare da apripista anche per altre malattie caratterizzate dallo stesso problema “di accesso”: per questo è importante continuare a sostenere la ricerca, come fa la Fondazione Telethon».
*Sala D et al, “Therapeutic advantages of combined gene/cell therapy strategies in a murine model of GM2 gangliosidosis”. Molecular Therapy - Methods & Clinical Development, March 16, 2022.