
Per Maddalena crescere non è scontato

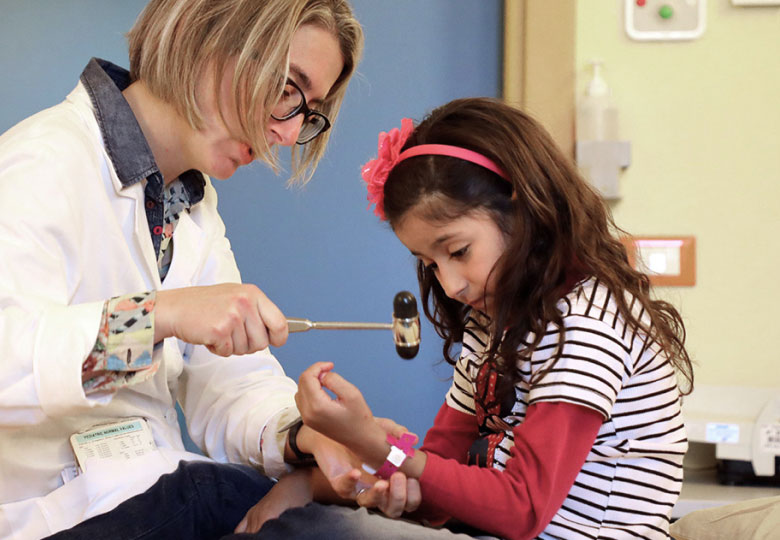
La storia di Maddalena inizia lontano, in Cina, ed è una storia fatta di piccole grandi conquiste, come andare a scuola, in bicicletta o nuotare. E con lei, sempre, mamma Valeria.



La storia di Maddalena inizia lontano, in Cina, ed è una storia fatta di piccole grandi conquiste, come andare a scuola, in bicicletta o nuotare. E con lei, sempre, mamma Valeria.
17.04.24
Scopriamo a che punto siamo nelle terapie dell'emofilia, il più frequente tra i disturbi della coagulazione.

17.04.24
Grazie allo strumento del Give at checkout di PayPal, sostenere la ricerca sulle malattie genetiche rare è ancora più semplice e veloce.
16.04.24
Dal 2 al 5 maggio a Padova i grandi esperti di scienza, cultura e innovazione a confronto. Fondazione Telethon content partner dell’edizione 2024 della manifestazione che promette di approfondire le nuove sfide di robotica, industry 4.0, space economy, life sciences e innovazione con oltre 50 eventi.

Con una donazione puoi davvero fare la differenza aiutando a crescere altri bambini con una malattia genetica rara come Federico.
Il tuo tempo può cambiare il futuro di chi affronta una malattia rara. Aiutaci a distribuire i nostri prodotti solidali nella modalità che preferisci per sostenere la ricerca.
